高通量短读比对工具
在过去的十几年里,随着高通量测序(HTS)成本降低,出现了各种测序概念, DNA-Seq, ChIP-Seq, RNA-Seq, BS-Seq覆盖了研究领域的方方面面。随之而来的问题是,如何把这些短片段快速且准确地回贴到参考基因组上。
解决这个问题不能直接使用传统的比对工具,比如说BLAST,因为它们的任务是找到最多的联配,而短序列比对工具则是要快速从众多潜在可选联配中找到最优的位置。也就是说BLAST和短读工具的目标其实不太一样。
在将海量的reads回贴到参考基因组上的过程,大量短读比对工具就需要面对准确度(accuracy)和精确度(precision)的平衡,也就是尽可能保证每一次的分析结果是相近的,并且也是符合真实情况。
mapping and alignment
对于alignment和mapping,其实我对他们之前的区别一直都不太清楚,并且也不知道它们到底该如何翻译,总感觉这两个词说的是同一件事情。这里看下Heng Li是如何进行定义
Mapping(映射)
- A mapping is a region where a read sequence is placed
- A mapping is regarded to be correct if it overlaps the ture region
Alignment(联配)
- An alignment is the detailed placement of each base in a read.
- An alignment is regarded to be correct if each base is placed correctly.
也就是说mapping侧重于把序列放到正确的位置,而不管这个序列的一致性,而联配则是主要让序列和参考序列尽可能的配对,而不管位置。目前来看,大多数工具都是想既能找到正确的位置,也保证有足够多的联配,不过明白这两者的区别对于不同项目的分析非常重要。比如说变异检测就要优先保证联配,而RNA-Seq则要尽可能保证把reads放到正确的位置。
如何挑选合适的短读比对工具
2012年 Bioinformatics 有一篇文章^[Tools for mapping high-throughput sequencing data ]综述了目前高通量数据的比对软件,并且建立主页https://www.ebi.ac.uk/~nf/hts_mappers/罗列并追踪目前的比对软件。
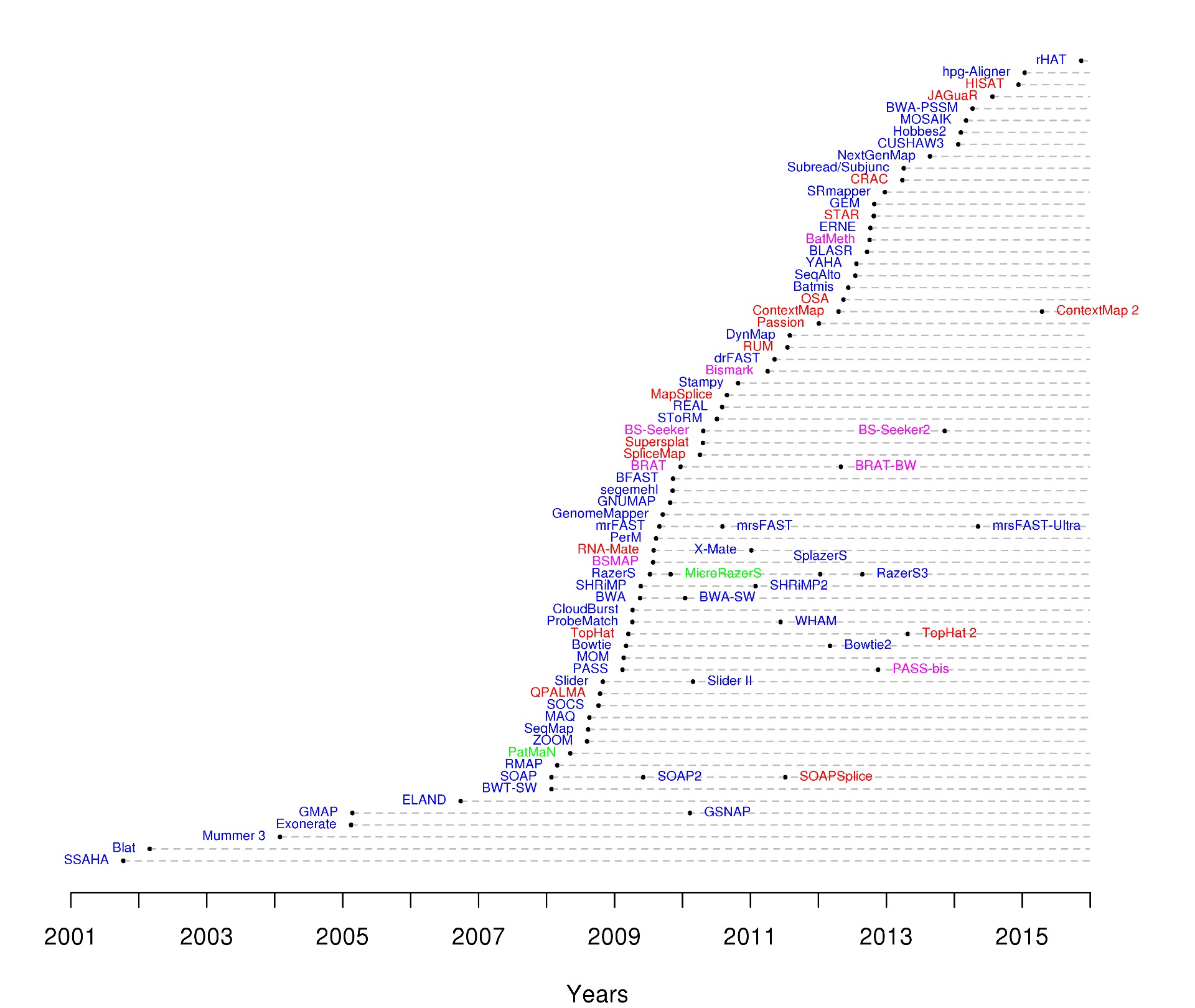
尽管看起来有那么多软件,但是实际使用就那么几种,BWA(傲视群雄), TopHat(尽管官方都建议用HISAT2,还是那么坚挺), SOAP(架不住华大业务多)。 由于这些工具都挺成熟,所以选择软件更多靠的是信仰,比如说Broad Institute的科学家喜欢bwa(毕竟是自家的),华大(BGI)喜欢用novoalign(也是自家出品),只不过novoalign是商业工具,不买就只能用单核,因此限制了它的传播。
除了信仰之外,我们挑选短序列比对工具的时候还要看什么呢?
- 联配算法: 全局,局部还是半全局
- 需要报道非线性重排(non-linear arrangements)嘛
- 比对工具如何处理InDels
- 比对工具支持可变剪切嘛
- 比对工具能够过滤出符合需要的联配嘛
- 比对工具能找到嵌合联配(chimeric alignments)嘛
最后我们的选择就落到两个工具:BWA和Bowtie2.
BWA和Bowtie的使用简介
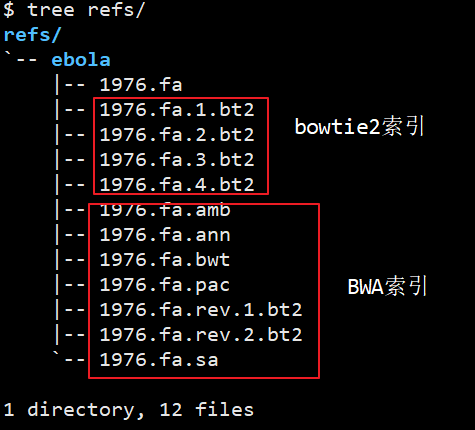
大部分比对工具的使用都可以分为两步,建立索引和比对索引。值得注意的是BWA有两种算法,aln和mem分别处理低于100bp和大于70bp的短读。bowtie也有1和2两代,处理50bp以下和50bp以上的短读,注意选择。
建立索引
需要先用efetch下载ebola参考基因组,如果网络不佳,直接去NCBI查找到下载也可以
mkdir -p ~/biostar/refs/ebola
cd ~/biostar
# efetch下载
efetch -db=nuccore -format=fasta -id=AF086833 > ~/refs/ebola/1976.fa
# wget下载
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/848/505/GCF_000848505.1_ViralProj14703/GCF_000848505.1_ViralProj14703_genomic.fna.gz
由于基因组特别小,所以建立索引的速度也会特别快。
REF=~/biostar/refs/ebola/1976.fa
# bwa
bwa index $REF
# bowtie2
bowtie2-build $REF $REF
序列比对
为了比对序列,首先得准备数据文件,可以从SRA上下载项目的Ebola项目的所有runs, 选择其中一个作为demo数据。
esearch -db sra -query PRJNA257197 | efetch -format runinfo > runinfo.csv
mkdir raw_data
cd raw_data
fastq-dump -X 10000 --split-files SRR1972739
比对其实很简单,如果只用默认参数的话
R1=raw_data/SRR1972739_1.fastq
R2=raw_data/SRR1972739_2.fastq
# bwa-mem
bwa mem $REF $R1 $R2 > bwa_mem_out.sam
# bowite2
bowtie2 -x $REF -1 $R1 -2 $R2 > bowtie2_out.sam
结果是个SAM文件,那什么是SAM呢,后面继续讨论。
bwa和bowtie2到底选谁
比较不同的比对软件是一个比较麻烦的事情。最常见的比较方法是,先模拟出一些序列,然后检查默认参数下的比对率和运行速度
- 10w条read,错误率为1%,默认参数
# dwgsim的安装方法见biostar handbook
~/bin/dwgsim -N 100000 -e 0.01 -E 0.01 $REF data
R1=data.bwa.read1.fastq.gz
R2=data.bwa.read2.fastq.gz
time bwa mem $REF $R1 $R2 > bwa.sam
# 4s 95.04%
time bowtie2 -x $REF -1 $R1 -2 $R2 > bowtie2.sam
# 10秒, 94.82%
- 10w条read,错误率为10%,默认参数
~/bin/dwgsim -N 100000 -e 0.1 -E 0.1 $REF data
R1=data.bwa.read1.fastq.gz
R2=data.bwa.read2.fastq.gz
time bwa mem $REF $R1 $R2 > bwa.sam
samtools flagstat bwa.sam
# 7s 83.16%
time bowtie2 -x $REF -1 $R1 -2 $R2 > bowtie2.sam
# 4秒,29.01%
在默认参数下,bowtie2的运行结果真的是差异巨大,尤其是10%的错误率下,几乎没有啥能够比对上了,让我们不禁怀疑bowtie2这个软件是不是不好使。
让我们换其他参数试试看
bowtie2 --very-sensitive-local -x $REF -1 $R1 -2 $R2 > bowtie.sam
# 10s, 63.21%
time bowtie2 -D 20 -R 3 -N 1 -L 20 -x $REF -1 $R1 -2 $R2 > bowtie.sam
# 11s, 87.11%
bowtie2在我们更换参数后比对率有着明显的提高,但是-D 2O -R 3 -N 1 -L 20如何得来呢?
也就是说bwa的默认参数是经过很好的优化来保证在默认参数下的结果,是不是我们都要选择bwa呢?也不能如此绝对,毕竟bowtie2的SAM结果保留了更多的信息。
最后说一句,选择比对软件在初学者时期真的是全靠信仰。