基因组组装
基因组组装一般分为三个层次,contig, scaffold和chromosomes. contig表示从大规模测序得到的短读(reads)中找到的一致性序列。组装的第一步就是从短片段(pair-end)文库中组装出contig。进一步基于不同长度的大片段(mate-pair)文库,将原本孤立的contig按序前后连接,其中会调整contig方向以及contig可能会存在开口(gap,用N表示),这一步会得到scaffolds,就相当于supercontigs和meatacontigs。最后基于遗传图谱或光学图谱将scaffold合并调整,形成染色体级别的组装(chromosome).
目前基于二代测序的组装存在挑战:
- 全基因组测序得到的短读远远小于原来的分子长度
- 高通量测序得到海量数据会增加组装的计算复杂性,消耗更高的计算资源
- 测序错误会导致组装错误,会明显影响contig的长度
- 短读难以区分基因组的重复序列
- 测序覆盖度不均一,会影响统计检验和结果结果诊断
上述的问题可以尝试从如下角度进行解决
- 短读长度:可以通过提供更多样本,并且建库时保证位置足够随机
- 数据集大小: 使用K-mers算法对数据进行组装。assembler不再搜寻overlap,而是搜索具有相同k-mers的reads。但是k-mer算法相比较overlap-based算法,灵敏度有所欠缺,容易丢失一些true overlaps。关键在于定义K。注: K-mer表示一条序列中长度为k的连续子序列,如ABC的2-mer为AB,BC
- 测序错误: 必须保证测序结果足够正确, 如提高质量控制的标准
- 基因组重复区: 测序深度要高,结果要正确。如果repeat短于read长度,只要保证有足够多且特异的read。如果repeat长于read,就需要paired ends or “mate-pairs”
- 覆盖度不均一: 提高深度,保证随机
- 组装结果比较:contig N50, scaffold N50, BUSCO
二代数据组装的算法和工具
基因组组装的组装工具主要分为三类:基于贪婪算法的拼接方法,基于读序之间的重叠序列(overlapped sequence)进行拼接的OLC(Overlap-Layout-Consensus)拼接方法和基于德布鲁因图(de bruijn graph)的方法,这三种方法或多或少基于图论。第一种是最早期的方法,目前已被淘汰,第二种适用于一代测序产生长片段序列,可以称之为字符串图(string graph),第三种是目前二代测序组装基因组的工具的核心基础,也就是要继续介绍的de bruijn图。
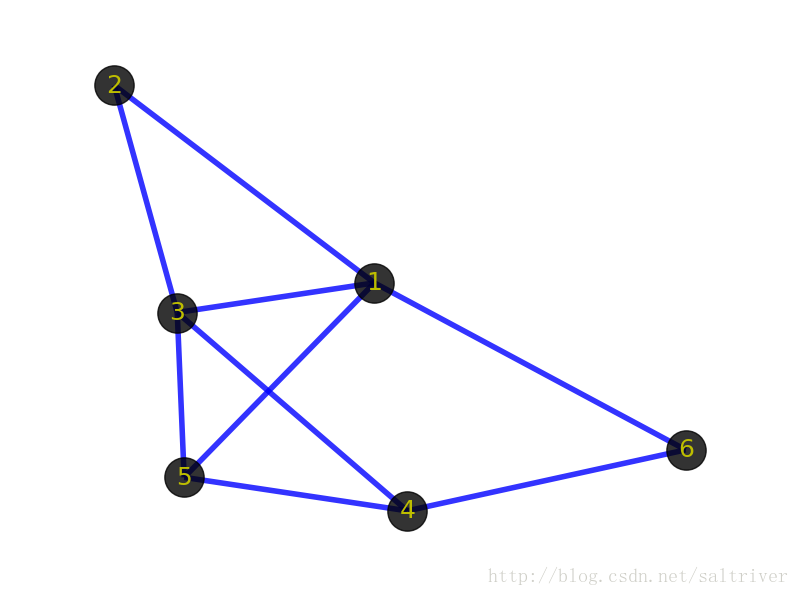
de bruijn图由两部分组成,节点(Nodes)和边(Edges),节点由k-mers组成,节点之间要想形成边就需要是两个k-mers存在K-1个完全匹配。比如说,ACTG, CTGC, TGCC在K=3时的k-mers为ACT,CTG,TGC,GCC,可以表示为ACT -> CTG -> TGC -> GC.
对于de brujin图而言,冗余序列不会影响k-mers的数量,比如说ACTG,ACTG,CTGC,CTGC,CTGC,TGCC,TGCC在K=3时依旧表示为ACT -> CTG -> TGC -> GCC。
上面是理想情况,实际序列中的测序错误,序列之间的SNP以及基因组低复杂度(重复序列)就会出现如下de brujin图
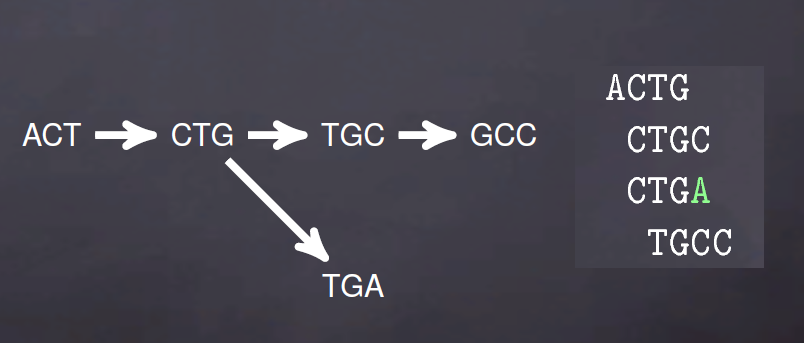
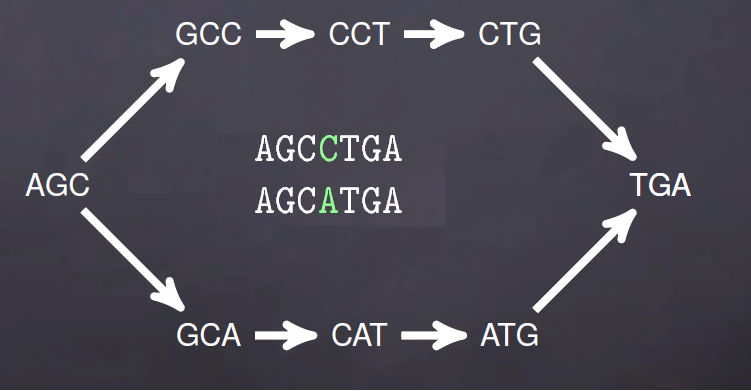
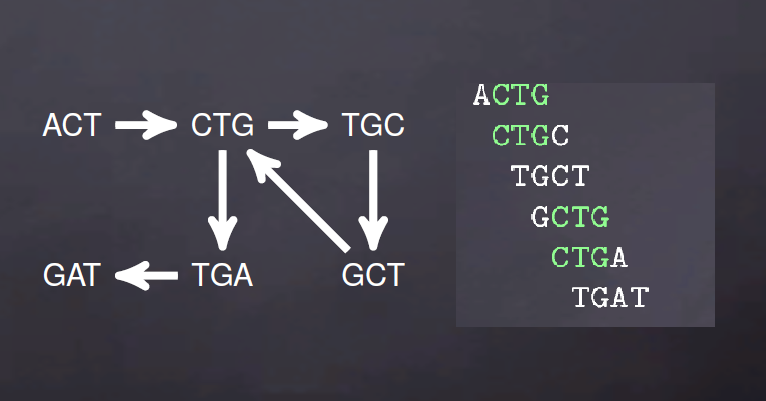
用图的方式表示就是下面情况
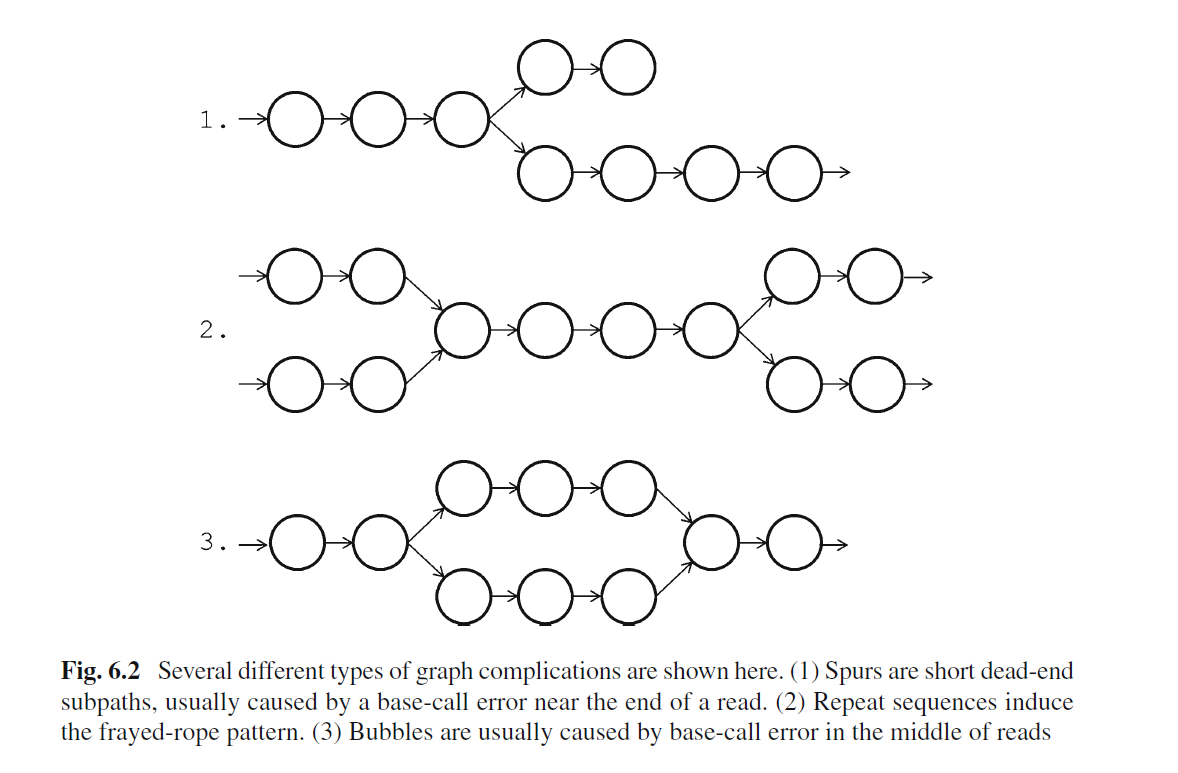
组装软件的任务就是从k-mers形成的图按照一定的算法组装出可能的序列,根据"GAGE: A critical evaluation of genome assemblies and assembly algorithms"以及自己的经验,目前二代数据比较常用的工具有Velvet, ABySS, AllPaths/AllPaths-LG, Discovar, SOAPdenovo, Minia, spades,Genomic Assemblers这篇文章有比较好的总结,
- ALLPaths-LG是公认比较优秀的组装工具,但消耗内存大,并且要提供至少两个不同大小文库的数据
- SPAdes是小基因组(<100Mb)组装时的首选
- SOAPdenovo是目前使用率最高的工具(华大组装了大量的动植物基因组),效率也挺好,就是错误率也高
- Minia是内存资源最省的工具,组装人类基因组contig居然只要5.7G的RAM,运行23小时,简直难以相信。
当然工具之间的差别并没有想象的那么大,也没有想象中那么小,可能在物种A表现一般的工具可能在物种B里就非常好用,因此要多用几个工具,选择其中最好的结果。
数据准备
这里使用来自于GAGE的金黄色葡萄球菌 Staphylococcus aureusa 数据进行练习。一方面数据量小,服务器能承受并且跑得快,另一方面本身基因组就组装的不错,等于是考完试能够自己对答案。
mkdir Staphylococcus_aureu && cd Staphylococcus_aureus
mkdir genome
curl ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/013/425/GCF_000013425.1_ASM1342v1/GCF_000013425.1_ASM1342v1_genomic.fna.gz > genome/Saureus.fna.gz
mkdir -p raw-data/{lib1,lib2}
curl http://gage.cbcb.umd.edu/data/Staphylococcus_aureus/Data.original/frag_1.fastq.gz > raw-data/lib1/frag_1.fastq.gz
curl http://gage.cbcb.umd.edu/data/Staphylococcus_aureus/Data.original/frag_2.fastq.gz > raw-data/lib2/frag_2.fastq.gz
curl http://gage.cbcb.umd.edu/data/Staphylococcus_aureus/Data.original/shortjump_1.fastq.gz > raw-data/lib2/shortjump_1.fastq.gz
curl http://gage.cbcb.umd.edu/data/Staphylococcus_aureus/Data.original/shortjump_2.fastq.gz > raw-data/lib2/shortjump_2.fastq.gz
基因组survey
在正式组装之前,需要先根据50X左右的illumina测序结果对基因组进行评估,了解基因组的大小,重复序列含量和复杂度。基于这些信息,确定后续策略以及是否真的需要对该物种进行测序。
基因组survery的核心就是使用k-mers对整体进行评估,k-mers时基因组里长度为k的子序列,当k=17时,ATCG的组合数就有170亿种,也就说理想条件下基因组大小只有超过17Gb才会出现2条一摸一样的k-mers。比如说有一个长度为14的序列,给定k-mers的k为8,于是能产生7条长度为8的子序列,于是推测基因组大小为7bp,但是这似乎和实际的14bp偏离有点远.
GATCCTACTGATGC (L=14), k-mers for k=8
n = (L-k) + 1 = 14 - 8 + = 7
GATCCTAC, ATCCTACT, TCCTACTG, CCTACTGA, CTACTGAT, TACTGATG, ACTGATGC
如果基因组大小为1MB, 那么当k-mers的k=18时,会得到(1000000-18)+1=999983个不同的k-mers,与实际大小偏差仅仅只有0.0017%,也就说基因组越大,预测就越接近。这是对单条基因组的估计结果,实际上高通量测序会得到基因组30X到50X深度的测序结果,比如说10个拷贝(C)的“GATCCTACTGATGC”在k-mers=8时会有70条子序列,
n = [(L-K) + 1] * C = [(14-8)+1]*10 = 70
为了得到实际的基因组大小,既需要将70除以拷贝数10,那么就得到了和之前一样的预测值7。当然上述都是理想条件,实际上测序不均一,低复杂区域,重复序列等都会影响预测结果。举个例子,"Genome sequencing reveals insights into physiology and longevity of the naked mole rat"的k=17, k_num=52,143,337,243,测序程度可以通过k-mers深度分布曲线来估计
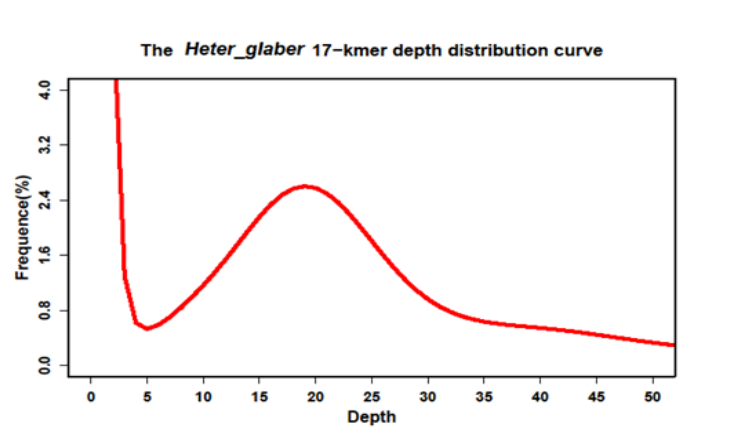
图中,深度为1的k-mers所占比例最高,表示绝大多数的k-mers仅仅出现了几次,这可能是测序错误造成。后续在depth=20逐渐形成一个峰,说明测序测度大概是20x附近,实际上是19x有极大值。于是基因组的大小就是"52,143,337,243/19=2744386170", 差不多就是2.74Gb
k-mers一般选择17即可,对于高度重复基因组或者基因组过大,可以选择19甚至31也行。但不是越大越好,因为如果一条reads里有一个错误位点,越大的k-mers就会导致包含这个错误位点的k-mers个数增多。
根据上述的介绍,便可以使用jellyfish统计k-mer,然后用R作图对基因组进行评估。当然这类工具其实已经有人开发,比如说ALLPATHS-LG/FindErrors,它不但能够修正低质量的短读,还能初步评估基因组,还有GCE(genome characteristics Estimation),由华大基因开发出来的一款基因组评估工具等。为了避免重复造轮子,简单就用这些工具即可。
使用GCE评估基因组: 先用kmer_freq_hash统计k-mer频数
# Staphylococcus_aureus项目根目录下
mkdir genome_survey && cd genome_survey
## 提供用于read的位置信息
ls raw-data/lib1/frag_*.fastq.gz > genome_survey/reads.list
## k-mer_freq_hash统计
~/opt/biosoft/gce-1.0.0/kmerfreq/kmer_freq_hash/kmer_freq_hash -k 15 -l genome_survey/reads.list -t 10 -o 0 -p genome_survey/sa &> genome_survey/kmer_freq.log
k-mer_freq_hash运行结束后会有粗略估计基因组大小,粗略估计为4.22Mb。注意,Kmer_individual_num 数据用于gce的输入参数。
随后用gce程序基于前面的输出结果进行估计
~/opt/biosoft/gce-1.0.0/gce -f genome_survey/sa.freq.stat -c 16 -g 108366227 -m 1 -D 8 -b 0 > genome_survey/sa.table 2> genome_survey/sa.log
# -c为主峰对应depth
# -g使用的就是Kmer_individual_num对应值
# -m 选择估算模型,真实数据选择1,表示连续型
在这次的日志文件中有预测后的结果4.34Mb,但是根据NCBI的数据,这个物种的基因组大小是2.8M左右。因此使用k-mers通过数学方法预测存在一定的局限性,需要结合流式细胞仪和粗组装的结果。
虽然也可以使用FindErrors对基因组进行评估,但是我实际使用时出现了各种问题,这里不做介绍。其他的工具也是大同小异,不做额外推荐。
基因组正式组装
当你拿到测序数据后,就可以按照如下几步处理数据。第一步是数据质控控制,这一步对于组装而言非常重要,处理前和处理后的组装结果可能会天差地别;第二步,根据经验确定起始参数,如K-mer和覆盖率;第三步,使用不同软件进行组装;第四步,评估组装结果,如contig N50, scaffold N50, 判断是否需要修改参数重新组装。
原始数据质量控制
尽管目前的测序技术已经非常成熟,公司提供的数据一般都可以直接用于普通的项目(特殊项目如miRNA-seq除外)。但由于 de novo 组装对数据质量比较敏感,因此需要通过质控来降低偏差。原始数据质量控制分为四个部分:
- 了解数据质量: 了解质量这一步可以暂时忽略,基本上基因组测序的结果都能通过FastQC的标准。
- 去接头和低质量reads过滤: 去接头和低质量reads过滤可供选择的软件非常之多,如NGSQCToolkit, Trimmomatic, cutadapter, 似乎都是国外开发的软件,但其实国内也有一款很优秀的工具叫做fastp
- 去除PCR重复: 去重一般都是在比对后根据位置信息进行,没有基因组的话只能根据PE的reads是否完全一样进行过滤。从理论上说,测序相当于是从基因组上随机抽样,不太可能存在完全一摸一样的两条序列。不过貌似只有FastUniq能做这件事情,后来有一个人写了sequniq。
- reads修正: 除了过滤或修剪低质量的reads外,一般而言,还需要对reads中的错误碱基进行修正。尤其当测序的覆盖度比较高时,错误的reads也就越来越多,会对 de novo 组装造成不良的影响。工具有BLESS2, BFC, Musket等,其中BLESS2的效率最高,效果也不错。
去接头和低质量reads过滤这一步推荐fastp,主要是因为它基于C/C++,运行速度块。
# 使用, 项目文件夹下
mkdir -p clean-data{lib1,lib2}
~/opt/biosoft/fastp/fastp -i raw-data/lib1/frag_1.fastq.gz -I raw-data/lib1/frag_2.fastq.gz -o clean-data/lib1/frag_1.fastq.gz -O clean-data/lib1/frag_2.fastq.gz
效果非常的惊人,直接干掉了90%的reads,从原来的1,294,104条变成77,375,一度让我怀疑软件是否出现了问题,直到我用同样的代码处理现在Illumina的测序结果以及看了FastQC的结果才打消了我的疑虑,没错,以前的数据质量就是那么差。注,除非是去接头,否则不建议通过删除序列的方式提高质量。
质控另一个策略是对短读中一些可能的错误碱基进行纠正,测序错误会引入大量无意义的K-mers,从而增加运算复杂度。此处使用BFC对测序质量:
~/opt/biosoft/bfc/bfc -s 3m -t 16 raw-data/lib1/frag_1.fastq.gz | gzip -1 > clean-data/lib1/corrected_1.fq.gz
~/opt/biosoft/bfc/bfc -s 3m -t 16 raw-data/lib1/frag_2.fastq.gz | gzip -1 > clean-data/lib1/corrected_2.fq.gz
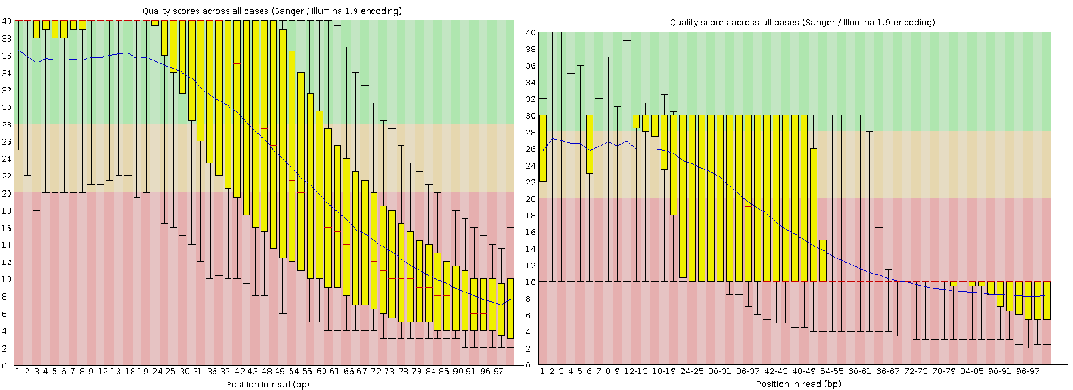
总之,质控的目标是在不引入的错误的情况下尽量提高整体质量,这一步对后续的组装影响很大,所以尽量做这一步,除非组装软件要求你别做,那你就不要手贱了。
使用不同工具和参数进行组装
二代组装可供选择的工具很多, 但是主流其实就那么几个, 所以组装的时候选择3~5个工具运行比较结果即可,比如说MaSuRCA
, IDBA-UD, SOAPdenovo2, Abyss, velvet和Spades。当然一旦你选择一个软件准备运行的时候,你就会遇到参数选择问题,比如怎么确定k-mers,组装软件最基础也是最核心的参数。这里有几条原则值得借鉴:
- k要大于log4(基因组大小),如果数学不好,无脑选择20以上
- 尽量减少测序错误形成的k-mers, 因为这是无意义的噪音, 也就是要求k不能过大
- 当然k也不能太小,否则会导致重复压缩,比如说ATATATA,在2kmers的情况下,就只有AT了
- 测序深度越高,K值也就可以选择的越大
但是说了那么多,你依旧不知道应该选择什么样的K,如果你的计算资源无限,那么穷举法最简单粗暴。如果穷举法不行,那么建议先用k=21, 55,77 组装一下contig, 对不同参数的contig N50有一个大致的了解,然后继续调整。此外还有一个工具叫做KmerGenie可以预测一个初始值。总之,让我们先运行第一个工具--SPAdes,可通过bioconda安装。
SPAdes全称是圣彼得堡基因组组装工具,包含了一系列组装工具处理不同的项目,如高杂合度的dipSPAdes,宏基因组的metaSPAdes。官方文档中以大肠杆菌为例运行整个流程,花了将近1个小时。我们的数据集比较小,速度会更快
# 项目根文件夹下
mkdir assembly/spades
spades.py --pe1-1 raw-data/lib1/frag_1.fastq.gz --pe1-2 raw-data/lib1/frag_2.fastq.gz --mp1-1 raw-data/lib2/shortjump_1.fastq.gz --mp1-2 raw-data/lib2/shortjump_2.fastq.gz -o assembly/spades/
你会发现之前说的k-mers在这里根本没出现,而且用的也是原始数据,这是因为spades.py有一个组件BayesHammer处理测序错误,并且它是多K类组装工具(multi-k assembly), 也就是说它会自动选择不同的K运行,从而挑选比较合适的k值,当然你还可以自己设置,比如说-k 21,55,77。最后结果为纠正后的短读数据,组装后的contig, 组装后的scaffold, 不同格式的组装graph。
同样运行多k-mers运行后比较的工具还有IDBA,它也有一系列的工具。IDBA是基础版,IDBA-UD适用于宏基因组和单细胞测序的数据组装,IDBA-Hybrid则是基于相似的基因组提高组装结果,IDBA-Tran是专门处理转录组数据。对于无参考基因组组装,作者推荐使用IDBA-UD。
IDBA-UD工具要求将两个配对的短读文件合并成一个,我们的原始数据需要先用它提供的fq2fa先转换格式
# 项目文件夹下
mkdir -p assembly/idba_ud
~/opt/biosoft/idba/bin/fq2fa --merge <(zcat clean-data/lib1/corrected_1.fq.gz) <(zcat clean-data/lib1/corrected_2.fq.gz) assembly/idba_ud/lib1.fa
~/opt/biosoft/idba/bin/fq2fa --merge <(zcat clean-data/lib2/corrected_1.fq.gz) <(zcat clean-data/lib2/corrected_2.fq.gz) assembly/idba_ud/lib2.fa
idba_ud和k-mers相关参数为--mink,--maxk,--step, 通过--read_level_x 传入不同大小的文库,也提供了短读纠正的相关参数--no_correct,--pre_correction
~/opt/biosoft/idba/bin/idba_ud -r assembly/idba_ud/lib1.fa --read_level_2 assembly/idba_ud/lib2.fa -o assembly/idba_ud/ --mink 19 --step 10
运行结束后在assembly/idba_ud下会生成一系列的文件,其中结果文件为contig.fa和scaffold.fa。
最后介绍一个要手动运行不同k-mers的工具,如ABySS, 它有一个亮点,就是能够可以使用多个计算节点。我们使用k=31进行组装
mkdir -p assembly/abyss
# 增加 /1,/2
sed 's/^@SRR.*/&\/1/' <(zcat raw-data/lib2/shortjump_1.fastq.gz) | gzip > raw-data/lib2/s1.fq.gz
sed 's/^@SRR.*/&\/2/' <(zcat raw-data/lib2/shortjump_2.fastq.gz) | gzip > raw-data/lib2/s2.fq.gz
~/opt/biosoft/abyss-2.0.2/bin/abyss-pe -C assembly/abyss k=31 n=5 name=asm lib='frag short' frag='../../raw-data/lib1/frag_1.fastq.gz ../../raw-data/lib1/frag_2.fastq.gz' short='../../raw-data/lib2/s1.fq.gz ../../raw-data/lib2/s2.fq.gz' aligner=bowtie
注意,首先ABYSS要求双端测序的reads命名要以/1和/2结尾,其次第二个文库才37bp, 所以比对软件要选择bowtie,否则你运行一定会遇到
histogram xxx.hist is empty的报错。当然到最后,这个问题我都没有解决掉,所以我放弃了。
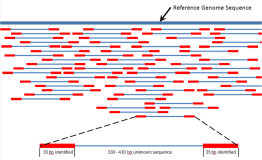
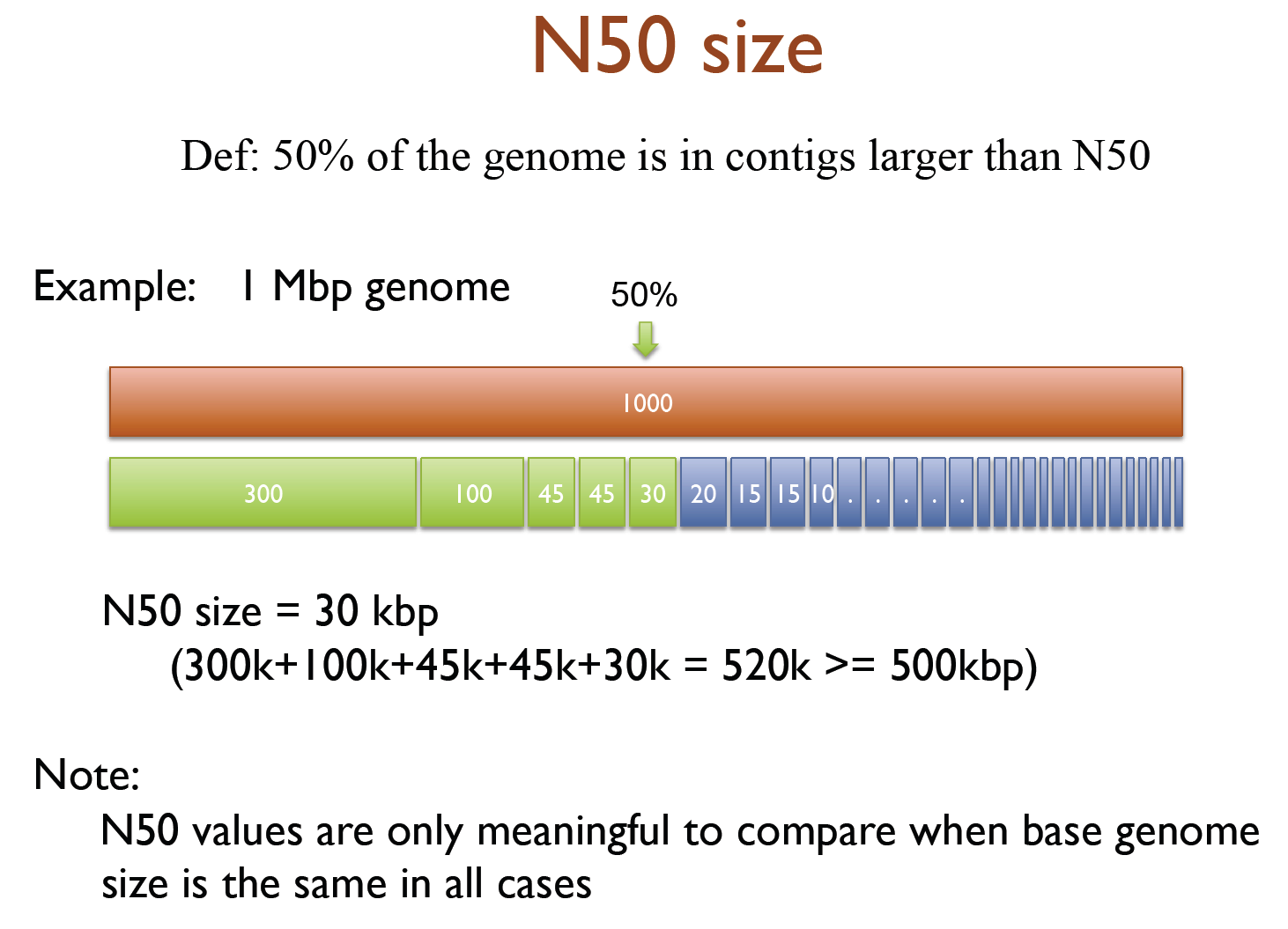
虽然看起来abyss用起来很简单,但其实背后的工作流程还是比较复杂,如下是它的流程示意图
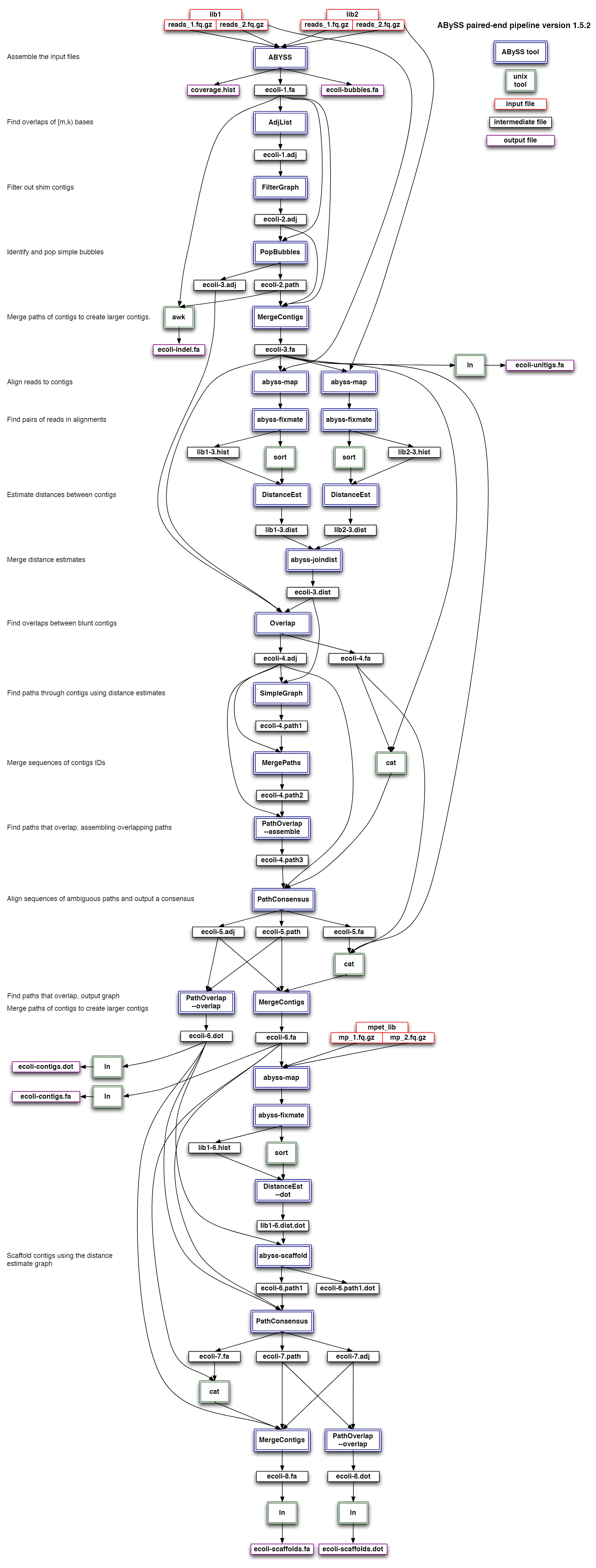
小结一下,这里用到了spades, idba,abyss三种工具对同一种物种进行组装,得到对应的contig结果,重点在于k-mers的选择。contig是组装的第一步,也是非常重要的一步,为了保证后续搭scaffold和基因组补洞等工作的顺利,我们先得挑选一个比较高质量的contig。
组装可视化和评估
理想条件下,我们希望一个物种有多少染色体,结果最好就只有多少个contig。当然对于二代测序而言,这绝对属于妄想,可以通过一款graph可视化工具bandage来感受一下最初得到的contig graph是多么复杂。
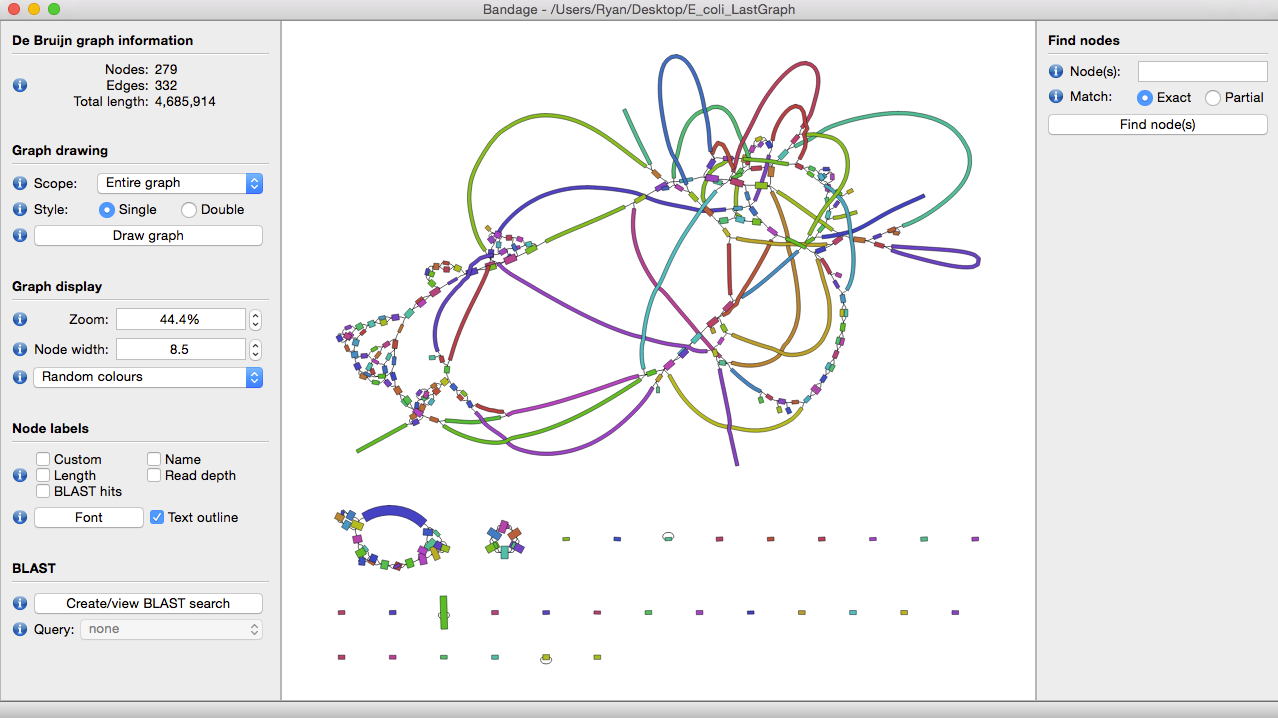
一般看这图直观感受就是怎么那么多节,这些节就是造成contig不连续的元凶。不同组装工具在构建de bruijn graph的差异不会那么大,contig的数量和大小和不同工具如何处理复杂节点有关。我们希望得到的contig文件中,每个contig都能足够的长,能够有一个完整的基因结构,归纳一下就是3C原则:
- 连续性(Contiguity): 得到的contig要足够的长
- 正确性(Correctness): 组装的contig错误率要低
- 完整性(Completeness):尽可能包含整个原始序列
但是这三条原则其实是相互矛盾的,连续性越高,就意味着要处理更多的模糊节点,会导致整体错误率上升,为了保证完全的正确,那么就会导致contig非常的零碎。此外,这三条原则也比较定性,我们需要更加定量的数值衡量,比如说contig数, 组装的总长度等, N50等。问题来了,什么叫做N50呢,
N50定义比较绕口,有一种只可意会不可言传的感觉,所以索性看图

假设一个基因组的大小为10,但是这个值只有神知道,你得到的信息就是组装后有3个contig,长度分别为"3,4,1,1",所以组装总长度为9。为了计算N50,我们需要先把contig从大到小排列,也就是"4,3,1"。然后先看最大的contig,长度是4,他的长度是不是超过组装总大小的一半了吗?如果是,那么N50=4, 4 < 4.5, 不是。 那么在此基础上加上第二长的contig,也就是4+3=7, 是不是超过一半了?7>4.5, 那么N50=3. 因此,N50的定义可以表述为"使得累加后长度超过组装总长度一半的contig的长度就是N50"。为了方便管理和使用软件,建议建立如下几个文件夹
N50是基于一个未知的基因组得到得结果,如果基因组测序比较完整,那么就可以计算NG50,也就是"使得累加后长度超过基因组总长度一半的contig的长度就是NG50"。NA50比较稍微复杂,需要将组装结果进一步比对到参考基因组上,以contig实际和基因组匹配的长度进行排序计算。
说完N50,我们介绍两款工具,QUAST和BUSCO。
QUAST使用质量标准(quality metrics)来评估不同组装工具和不同参数的组装效果,无论是否有基因组都可以使用。我们分别以有参和无参两种模式比较Minia,IDBA和SPAdes三个组装的运行结果
# without reference
quast.py -o compare idba_ud/contig.fa minia/minia.contigs.fa spades/contigs.fasta
# with reference
quast.py -R ../genome/Saureus.fna -o compare idba_ud/contig.fa minia/minia.contigs.fa spades/contigs.fasta
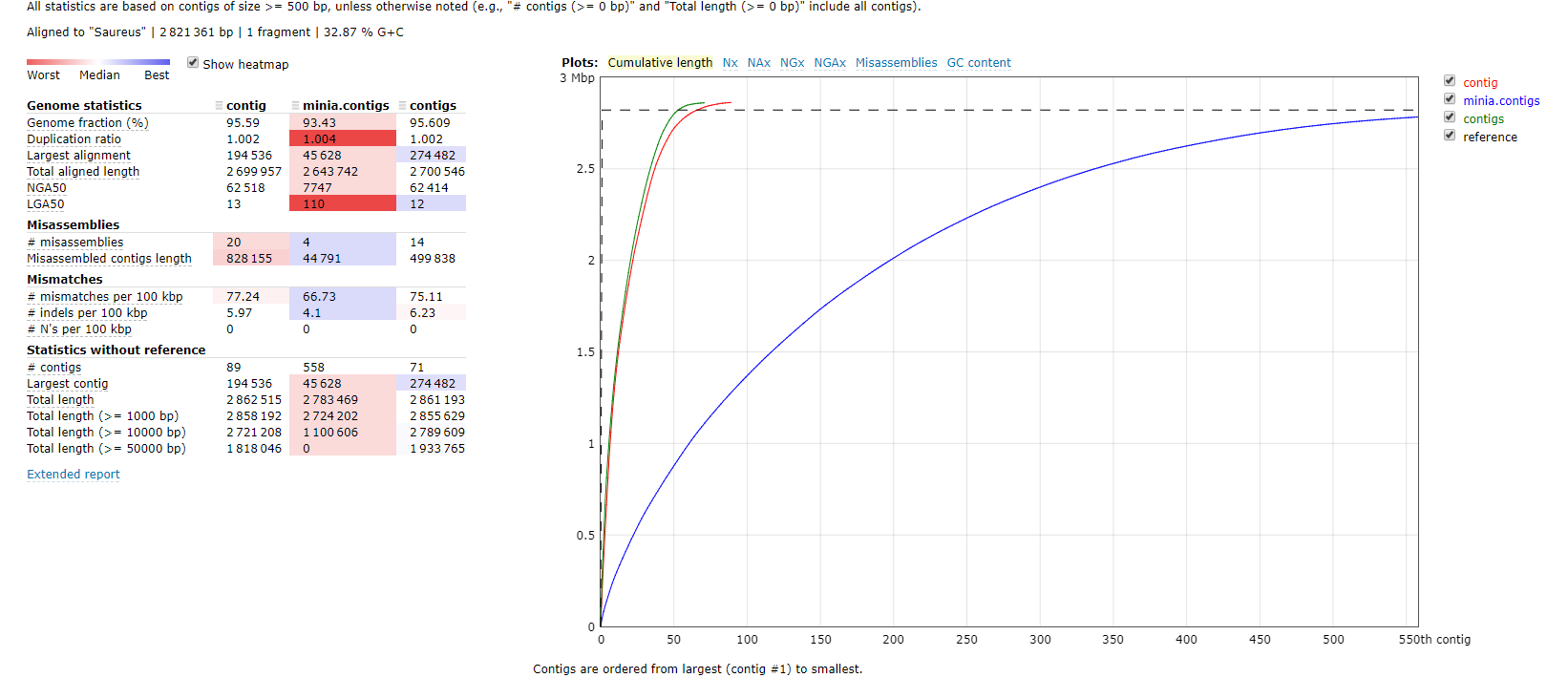
这个结果非常直观的告诉我们一个事实就是spades组装的contigs`各方面表现都很优秀,minia由于内存使用率最低,所以组装效果一般也是可以理解。
BUSCO通过同源基因数据库从基因完整度来评价基因组组装结果。BUSCO首先构建了不同物种的最小基因集,然后使用HMMER,BLAST,Augustus等工具分析组装结果中的同源基因,从而定量评估组装是否完整。
busco -i assembly/spades/contigs.fasta -o result -l /home/wangjw/db/busco/bacteria_odb9 -m genome -f
运行结果会在当前目录下的run_result生成一些列文件,其中的short_summary_result.txt内容如下
# Summarized benchmarking in BUSCO notation for file assembly/spades/contigs.fasta
# BUSCO was run in mode: genome
C:98.6%[S:98.6%,D:0.0%],F:0.0%,M:1.4%,n:148
146 Complete BUSCOs (C)
146 Complete and single-copy BUSCOs (S)
0 Complete and duplicated BUSCOs (D)
0 Fragmented BUSCOs (F)
2 Missing BUSCOs (M)
C值表示和BUSCO集相比的完整度,M值表示可能缺少的基因数,D则是重复数。正所谓没有比较,就没有伤害,我们拿之前QUAST对比中表现比较差的minia结果作为对比。
C:85.1%[S:85.1%,D:0.0%],F:2.7%,M:12.2%,n:148
126 Complete BUSCOs (C)
126 Complete and single-copy BUSCOs (S)
0 Complete and duplicated BUSCOs (D)
4 Fragmented BUSCOs (F)
18 Missing BUSCOs (M)
98% vs 85%, 一下子对比就出来了。综上,从两个维度上证明的SPAdes不但组装效果好,而且基因完整度也高,当然它的内存消耗也是很严重。这都是取舍的过程。
附录
参考资料
软件安装
由于不同软件对不同的基因组的适合度不同,一般都需要参数多个工具的不同参数,根据N50和BUSCO等衡量标准选择比较好的结果。为了避免后续花篇幅在工具安装上,因此先准备后续的分析环境。对于组装而言,我们需要安装如下工具:
- 质量控制:
- FastQC
- fastp
- BFC
- 主流组装工具:
- ABySS
- IDBA
- SOAPdenovo2
- Velvet
- Sapdes
- Minia
- Ray
- MasuRCA
- 基因组组装评价工具
- BUSCO
- Quast
- 基因结构预测和功能注释暂时不在考虑范围内
以下操作所用服务器的基本信息为:Linux的内核为3.10.0-693.el7.x86_64, GCC版本为4.8.5。为了方便管理和使用软件,建议建立如下几个文件夹, 分门别类的存放不同工具及其源码。
# 普通用户
mkdir -p ~/opt/{sysoft,biosoft}
mkdir -p ~/src
# 管理员
sudo mkdir -p /opt/{sysoft,biosoft}
sudo mkdir -p /src
sudo chmod 1777 /opt/biosoft /opt/sysoft /src
系统自带的GCC版本是4.8,而BLESS2要求4.9+, ABySS要求6.0+,直接编译这些工具可能会出错,但直接升级系统的GCC版本可能会影响整体稳定性,因此推荐将在opt/sysoft下安装高版本的GCC。当然GCC的版本也不是越高越好,最好和作者开发的版本一致,也就是他们要求的最低版本。
# gcc,mpfr,gmp,mpc,isl
cd ~/src
wget -4 https://mirrors.tuna.tsinghua.edu.cn/gnu/gcc/gcc-6.4.0/gcc-6.4.0.tar.xz
tar xf gcc-6.4.0.tar.xz
cd gcc-6.4.0
./contrib/download_prerequisites
mkdir build && cd build
../configure --prefix=$HOME/opt/sysoft/gcc-6.4.0 --enable-threads=posix --disable-multilib --with-system-zlib
make -j 8 && make install
根据我之前关于GCC编译的文章,程序编译不成功大多是因为找不到头文件(存放在include目录下)和链接库文件(存放在lib目录下),默认编译头文件只会搜索/usr/include,/usr/local/include, 而链接库文件只会搜索/lib,/usr/lib[64],/usr/local/lib[64]. 为了让编译完成的GCC的头文件和链接库文件能被搜索到,需要在~/.bashrc文件中添加几个环境变量:
-
PKG_CONFIG_PATH: 同时添加搜索头文件和链接头文件的路径 -
C_INCLUDE_PATH: 编译时搜索头文件的路径 -
LIBRARY_PATH: 编译时搜索链接文件的路径 -
LD_LIBRARY_PATH: 运行时搜索链接文件的路径
即添加如下几行内容到~/.bashrc文件中,并执行source ~/.bashrc更新环境变量。
export PKG_CONFIG_PATH=~/opt/sysoft/gcc-6.4.0/lib64/pkgconfig:$PKG_CONFIG_PATH
export C_INCLUDE_PATH=~/opt/sysoft/gcc-6.4.0/include:$C_INCLUDE_PATH
export LIBRARY_PATH=~/opt/sysoft/gcc-6.4.0/lib64:$LIBRARY_PATH
export LD_LIBRARY_PATH=~/opt/sysoft/gcc-6.4.0/lib64:$LD_LIBRARY_PATH
export PATH=~/opt/sysoft/gcc-6.4.0/bin:$PATH
genome survey工具: 功能都类似,GCE安装最方便胜出
cd ~/src
wget ftp://ftp.genomics.org.cn/pub/gce/gce-1.0.0.tar.gz
tar xf gce-1.0.0.tar.gz -C ~/opt/biosoft
组装软件种类很多,对于小基因组(<100Mb)而言SPAdes是很好的选择, 但是对于大基因组就得多试试几个,比如说MaSuRCA, Discover de novo, Abyss,SOAPdenovo2, IDBA。内存不太够的话可以尝试Minia。
组装软件一:ABySS的安装依赖boost1.62, OpenMPI, Google/sparsehash, SQLite,且GCC支持OpenMP,因此也就是一个个下载,一个个安装的过程。
# boost1.62
cd ~/src
wget -4 https://sourceforge.net/projects/boost/files/boost/1.62.0/boost_1_62_0.tar.bz2
tar xf boost_1_62_0.tar.bz2
cd boost_1_62_0
./bootstrap.sh --prefix=$HOME/opt/sysoft/boost-1.62
./b2
# 引入头文件的路径为~/src/boost_1_62_0, 引入链接库的路径为~/src/boost_1_62_0/stage/lib
# openmpi
wget https://www.open-mpi.org/software/ompi/v3.0/downloads/openmpi-3.0.0.tar.gz
tar xf openmpi-3.0.0.tar.gz
cd openmpi-3.0.0
./configure --prefix=$HOME/opt/sysoft/openmpi-3.0.0
make -j 8 && make install
# 在.bashrc中添加环境变量或手动修改也行
echo 'export PKG_CONFIG_PATH=~/opt/sysoft/openmpi-3.0.0/lib/pkgconfig:$PKG_CONFIG_PATH' >> ~/.bashrc
echo 'export PATH=~/opt/sysoft/openmpi-3.0.0/bin:$PATH' >> ~/.bashrc
# sparsehash
cd ~/src
git clone https://github.com/sparsehash/sparsehash.git
cd sparsehash
./configure --prefix=$HOME/opt/sysoft/sparsehash
make && make install
# sqlite
cd ~/src
wget -4 http://www.sqlite.org/2018/sqlite-tools-linux-x86-3220000.zip
unzip sqlite-tools-linux-x86-3220000.zip
mv sqlite-tools-linux-x86-3220000 ~/opt/sysoft/sqlite3
最后在安装ABySS时要以--with-PACKAGE[=ARG]形式指定依赖软件的路径
cd ~/src
wget -4 http://www.bcgsc.ca/platform/bioinfo/software/abyss/releases/2.0.2/abyss-2.0.2.tar.gz
tar xf abyss-2.0.2.tar.gz
cd abyss-2.0.2
./configure --prefix=$HOME/opt/biosoft/abyss-2.0.2--with-boost=$HOME/src/boost_1_62_0 --with-sparsehash=$HOME/opt/sysoft/sparsehash --with-sqlite=$HOME/opt/sysoft/sqlite3
make && make install
组装软件二:SOAPdenovo2,华大出品,目前使用率最高的工具
cd ~/src
git clone https://github.com/aquaskyline/SOAPdenovo2.git
cd SOAPdenovo2
mkdir -p ~/opt/biosoft/SOAPdenovo2
mv SOAPdenovo-* ~/opt/biosoft/SOAPdenovo2/
组装软件三: IDBA. de Brujin图依赖于K-mers的k的选择,IDBA能够自动化递归使用不同的k进行组装,从而确定最优的K。
cd ~/src
git clone https://github.com/loneknightpy/idba.git
idba/build.sh
mv idba ~/opt/biosoft/
组装软件四:MaSuRCA,能够纯用二代,也能二代三代测序混合使用,先用 de bruijn 图构建长reads,然后再用OLC算法进行组装
cd src
wget ftp://ftp.genome.umd.edu/pub/MaSuRCA/latest/MaSuRCA-3.2.4.tar.gz
tar xf MaSuRCA-3.2.4.tar.gz
cd MaSuRCA-3.2.4
export DEST=$HOME/opt/biosoft/MasuRCA
./install.sh
质控软件一: 原本是要推荐BLESS2,但是这个软件在编译完成后出现各种核心转移的毛病,和我的系统相性太差,于是改用Li Heng的BFC
cd ~/src
git clone https://github.com/lh3/bfc.git
cd bfc
make
mkdir -p ~/opt/biosoft/bfc
mv bcf hash2cnt ~/opt/biosoft/bfc
质控软件二: fastp是一款基于C/C++编写的工具,速度会比较块,而且运行之后会有比较好看的图哦
mkdir -p ~/opt/biosoft/fastp
cd ~/opt/biosoft/fastp
wget http://opengene.org/fastp/fastp
chmod a+x ./fastp
评估工具一:Quast, 它通过比较N50,N G50等参数来评价基因组组装质量.Quast由Python编写,推荐使用bioconda安装
conda create --name assembly python=2.7
source activate assembly
conda install quast
评估工具二: BUSCO,这是一个利用进化信息从基因完整性角度评估组装准确性的工具,推荐使用biconda安装。
source activate assembly
conda install busco
尽管conda安装了busco,但是离实际运行还需要添加几个环境变量和不同物种的基因数据集,请使用printenv确保如下如下几个路径都已经添加到环境变量中。
export PATH="/path/to/AUGUSTUS/augustus-3.2.3/bin:$PATH"
export PATH="/path/to/AUGUSTUS/augustus-3.2.3/scripts:$PATH"
export AUGUSTUS_CONFIG_PATH="/path/to/AUGUSTUS/augustus-3.2.3/config/"
之后,根照自己研究的物种在http://busco.ezlab.org/选择进化上接近的评估数据集,比如说你如果研究鱼,那么"actinopterygii(辐鳍鱼类)"就比"metazoa(多细胞动物)"更加合适.
实际运行时可能还存在链接库无法找寻以至于程序出错,解决方法就是将相对应或着接近的库拷贝或软链接到
~/miniconda3/env/assembly/lib下。